MWAS practical
The data for todays practical can be downloaded from the Vital-It server using the command scp. It will need to be on your local machine:
$ scp username@prd.vital-it.ch:/scratch/cluster/monthly/kridout/summer2012_Day2.zip ./
To annotate our genome we will need an automated pipeline of repeat masking, homology searching and gene prediction. To do this we will be using the program MAKER on the webserver MWAS.
To avoid overloading the MWAS server, PLEASE WORK IN PAIRS AND SEND ONLY A SINGLE MAKER REQUEST.
Depending on the type of analysis to be performed we could try to annotate all scaffolds, or we might identify scaffolds of interest. Here we will pick a number of interesting scaffolds out of yesterdays assemblies. We will do this using BLAST.
First, we will need to login to Vital-It.
$ ssh username@prd.vital-it.ch
$ ssh dee-serv02.vital-it.ch
Make the directories that we will work in:
$ mkdir /scratch/cluster/weekly/username/MWAS
$ mkdir /scratch/cluster/weekly/username/MWAS/BLAST
$ mkdir /scratch/cluster/weekly/username/MWAS/BLAST/db
Change into the MWAS folder and collect a list of interesting proteins.
$ cd /scratch/cluster/weekly/username/MWAS
$ unzip /scratch/cluster/monthly/kridout/Interesting_genes.fa.zip
We need to temporarily add the BLAST executables to your $PATH (do not place this permanently into your .bashrc as you will need to run an older version of BLAST for another practical):
$ source /mnt/common/Blast/Blast.bashrc
Next we need to turn the scaffolds into a BLAST database for searching. First change into the BLAST directory and make the database:
$ cd /scratch/cluster/weekly/username/MWAS/BLAST
$ makeblastdb -in [path_to_your_best_scaffolds] -dbtype nucl -out [db/db_name]
Now we blast our interesting proteins against the scaffolds assembled yesterday to find those of interest.
$ tblastn -db [db/db name] -query [path_to_Interesting_genes.fa] -evalue 2e-8 -out [output name] -outfmt 6
Finally, we will run a script to make a fasta file of all the matching scaffolds.
$ perl /scratch/cluster/monthly/kridout/scaffoldsFromBlast.pl [path_to_your_best_scaffolds] [blast_output] > [scaffold_subset]
Use abyss-fac to see how many scaffolds you have collected and the range of scaffold lengths.
Exit Vital-It and Collect the file using scp.
$ scp username@prd.vital-it.ch:/scratch/cluster/weekly/username/BLAST/[scaffold_subset] ./
The remainder of this practical will be done on your local machine.
>Navigate to the MAKER website:
http://derringer.genetics.utah.edu/cgi-bin/MWAS/maker.cgi
>Make a new login using your username (i.e. student4) and select the New Job tab
The Denovo Annotation section should contain any sequences to be annotated. In our case, this means the scaffold subset that we have just collected.
>Upload your scaffold file
EST evidence from our study species or a relative can be added.
>Upload the transcript file Sinvicta_tr.fa from the day 2 files
Searching the full swissprot database and the full repeat database will take too much time, so a subset of proteins and repeats has been prepared for you.
>Upload the protein file Swissprot_subset.prot
>Upload the repeats file Sinvicta_repeats.fa
>Under Denovo Annotation select your scaffold file
>Under EST Evidence select the transcript file
>Under Protein Homology Evidence select the swissprot protein file
>Under the Configure Repeat Masking section select the repeats file
>Select the course training files for SNAP (we will not be using AUGUSTUS and GeneMark, although this is recommended)
Very short scaffolds might still contain whole genes, however there is a high likelihood of broken genes in these regions. In the interest of time and accuracy we will skip them.
>Under Annotation Properties set the minimum contig length to 1000
>Select Add Job to Queue to launch MAKER
This process will take several hours. You can monitor the status of your job from the Running Jobs tab.
MWAS Questions
You should have now started the process of genome annotation. Generating a set of reliable genes is not a trivial task. It is very important to understand the potential problems with genes predicted with automated methods. Consider the data that you have produced this far and answer the following questions:
Q1. Genome/transcript assembly does not yet produce scaffolds that are guaranteed to be accurate. What effects could misassemblies in the data have on genome annotation?
Q2. Errors in the EST or protein annotation databases will be perpetuated through this annotation process. This is a problem for all homology based annotation methods. How might you try to deal with this uncertainty?
Q3. What are the pros and cons of a homology based gene finding approach?
Q4. What are the pros and cons of a denovo prediction approach (e.g. AUGUSTUS) for gene finding?
Q5. What are the potential biases generated by predicting genes in this way?
WHILE MAKER IS RUNNING CHECK THAT YOU HAVE APPOLO INSTALLED AND FUNCTIONING
If Apollo is not installed, you should download and install it before we continue with the Apollo practical. Downloading it from the local server will be fastest.
Apollo manual gene editing
Sometimes we might want to view or manually edit specific genes. To perform this task, programs such as IGV or Apollo can be used.
>Download all data from MWAS (under View Results)
>Open Apollo and load the gff file (this is gff3 format) of all scaffolds downloaded from MAKER. Untick the box labeled Embedded sequence and select the original scaffold file
The top 2 panes represent the forwards strand and the bottom 2 are the reverse. Results from the different prediction methods are displayed in the black panes and the final annotations in the blue.
>Click on the squares (predictions) in the black panes to see which programs have produced the different results
>To color the different prediction methods, right click on the prediction (in the black panel) and select Change color of this feature type
Scaffold and gff3 files have been prepared for you to examine and compare with your own annotations. Open the fasta file Sinvicta_good_scaffolds.fa and the gff3 file Sinvicta_good_predictions.gff3 in Apollo.
Q6. What are the main differences that you see between the files (do not try to investigate individual genes)?
Q7. Based on these results, what can you say about which programs over predict and which under predict? What do you think of the reliability of these programs?
The gff3 file given to you contains examples of correctly and incorrectly annotated genes. In some places genes have been forced together, or separated at introns. Some genes also have incorrect splice sites.
Q8. Using Apollo, identify likely genes that might have been badly annotated. Why did you choose these genes?
Using tools such as Apollo we can perform manual editing of gene predictions. Splice sites occur at the intron/exon boundary and are highly conserved. Apollo highlights the ends of exons that do not have the necessary splicing sites using an orange triangle:
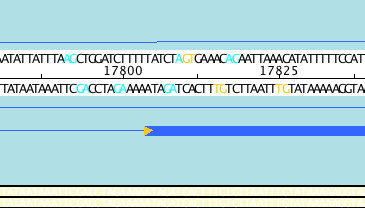
Acceptable splice sites can be colored as in the above example.
>Right click on the nucleotide sequence to select color by splice site potential (you will not see the nucleotides until you have zoomed in sufficiently far)
Simply choosing the closest splice site is not a reliable method for fixing this prediction.
Q9. How might you determine the true splice positions of a predicted gene?
The gene maker-Sign00006-snap-gene-0.10-mRNA-1 contains a number of incorrect splicing sites identified by Apollo.
>Select this gene under the Annotation tab for a better view and zoom in to examine the sequence
Q10. Do you think that this gene represents a real protein? Explain your answer. What else might this gene be?
A protein domain scan on the whole genome determined that the fire ant genome contains multiple Vitellogenins. This is potentially interesting because the single Vitellogenin of the honey bee plays roles in determining lifespan and division of labor (Amdam et al 2012). Protein2genome suggest there are three vitellogenins in our scaffold:
maker-Sign00006-snap-gene-1.41-mRNA-1
maker-Sign00006-snap-gene-1.42-mRNA-1
maker-Sign00006-snap-gene-1.43-mRNA-1
Q11. What evidence supports these predictions?
Gene maker-Sign00006-snap-gene-1.40-mRNA-1 is considerably shorter, but has also matched this protein. Experimental evidence suggests that there is indeed a 4th Vitellogenin gene, despite the missing EST evidence.
A closer examination of the BLAST evidence suggests that this gene has been truncated by the prediction software.
Q12. Why do you think this is?
We will attempt to extend this gene. Given more time we would examine the BLAST results against a number of Vitellogenin homologues to determine the correct splice, start and stop sites. Here, we will try to combine the BLASTX prediction and EST results.
First, we should duplicate the gene(s) that we intend to work on in case we make a mistake.
>Double click on each of these genes to select it as a whole. Right click on the selected gene and choose Duplicate transcript
The first new BLAST exon (from right to left) does not begin at a splice site, however this can be extended using EST evidence which does.
>Right click in the blue panel underneath the nucleotide that your new exon should start at. Select Create new annotation -> gene
>Set the new gene length to a single base
>Find the end of your exon and repeat this process
>Hold the shift key and click on the two new genes to select them both. Right click on one of these and select Merge transcripts
>Double click on the new gene to select the whole thing. Right click on this gene and select Merge exons
You have built your first new exon. Apollo will place an orange triangle at the end of the gene if the splice site was not correct.
>Check your gene for correct splicing and frame. If the frame is incorrect there will be stop codons.
>If your exon is correctly spliced, you should try to join it to the rest of the gene. Select the gene and the new exon, right click and Merge transcripts
>You may now find that the splicing does not work. This is because the original gene was built to end at a stop codon. Use the EST and BLAST evidence to find the correct splice site. Make sure that the correct reading frame is maintained
To move the end of the exon by small amounts, right click on the transcript and select Exon detail editor. Navigate to the necessary part of the sequence (remember that the sequence will be displayed from left to right, where as these genes on the reverse strand are viewed in the pale blue panel from right to left).
>Repeat this process until you reach the end of the gene. Remember to stop the gene at a stop codon.
Predicted genes:
maker-Sign00006-snap-gene-6.41-mRNA-1
maker-Sign00006-snap-gene-6.43-mRNA-1
Both map in parts to the same EST. It is possible that these genes have been fragmented by Apollo and belong to the same transcript.
Q13. How might you try to determine whether these are the same gene?
Extension
Despite possible errors in the databases, genes can be annotated using automated BLAST searching. A commonly used tool for automated annotation is BLAST2GO, which allows the user to annotate using a gene name, domain, function, ontology and pathway. This can be very useful for downstream processes.
If you have finished editing your genes, choose a subset (maximum 10) and explore the functionality of BLAST2GO: