Ants, Bees, Genomes & Evolution @ Queen Mary University London
Publications See also [Google Scholar] & [Software Tools]
Legend: *Joint first authors, °Corresponding authors, Authors from the Wurmlab
Categories: all primary research review-
Expression of subunits of an insecticide target receptor varies across tissues, life stages, castes, and species of social bees
A Witwicka*, F López-Osorio*, V Patterson, Y Wurm°
Global losses of insects jeopardize ecosystem stability and crop pollination. Robust evidence indicates that insecticides have contributed to these losses. Notably, insecticides targeting nicotinic acetylcholine receptors (nAChRs) have neurotoxic effects on beneficial insects. Because each nAChR consists of five subunits, the alternative arrangements of subunits could create a multitude of receptors differing in structure and function. Therefore, understanding whether the use of subunits varies is essential for evaluating and predicting the effects of insecticides targeting such receptors. To better understand how the use and composition of nAChRs differ within and between insect pollinators, we analyzed RNA-seq gene expression data from tissues and castes of Apis mellifera honey bees and life stages and castes of the Bombus terrestris bumble bees. We reveal that all analyzed tissues express nAChRs and that relative expression levels of nAChR subunits vary widely across almost all comparisons. Our work thus shows fine-tuned spatial and temporal expression of nAChRs. Given that co-expression of subunits underpins the compositional diversity of functional receptors and that the affinities of insecticides depend on nAChR composition, our findings provide a likely mechanism for the various damaging effects of nAChR-targeting insecticides on insects. Furthermore, our results indicate that the appraisal of insecticide risks should carefully consider variation in molecular targets.
Molecular Ecology, 2023 -
No supergene despite social polymorphism in the big-headed ant Pheidole pallidula
E Favreau, C Lebas, E Stolle, A Priyam, R Pracana, S Aron, Y Wurm
Ant colonies ancestrally contained one queen and her non-reproductive workers. This is also the case for many but not all colonies of the Mediterranean big-headed ant Pheidole pallidula. Indeed, this species also has a derived form of social organization with multiple reproductive queens in the colony. The co-existence of two social forms also independently evolved in three other lineages of ants. In each of those lineages, variants of a supergene region of suppressed recombination determine social form. This is likely because supergene regions can link advantageous combinations of alleles from multiple loci. We thus hypothesized that a supergene region also determines colony queen number in the big-headed ant. To test this, we performed extensive population genetic analyses and genomic comparisons. We find no evidence of a supergene-like region with differentiation between single- and multiple-queen colonies. Our results show that a complex social polymorphism can evolve and be maintained without supergenes.
bioRxiv 2022.12.06.519286 -
Convergent evolution of a labile nutritional symbiosis in ants
[DOI]
R Jackson, D Monnin, PA Patapiou, G Golding, H Helanterä, J Oettler, J Heinze, Y Wurm, CK Economou, M Chapuisat, LM Henry
Ants are among the most successful organisms on Earth. It has been suggested that forming symbioses with nutrient-supplementing microbes may have contributed to their success, by allowing ants to invade otherwise inaccessible niches. However, it is unclear whether ants have evolved symbioses repeatedly to overcome the same nutrient limitations. Here, we address this question by comparing the independently evolved symbioses in Camponotus, Plagiolepis, Formica and Cardiocondyla ants. Our analysis reveals the only metabolic function consistently retained in all of the symbiont genomes is the capacity to synthesise tyrosine. We also show that in certain multi-queen lineages that have co-diversified with their symbiont for millions of years, only a fraction of queens carry the symbiont, suggesting ants differ in their colony-level reliance on symbiont-derived resources. Our results imply that symbioses can arise to solve common problems, but hosts may differ in their dependence on symbionts, highlighting the evolutionary forces influencing the persistence of long-term endosymbiotic mutualisms.
The ISME Journal, Volume 16, pages 2114-2122, (2022) -
Individual-based Modeling of Genome Evolution in Haplodiploid Organisms
[DOI]
R Pracana, R Burns, R L Hammond°, B C Haller, Y Wurm°
Ants, bees, wasps, bark beetles, and other species have haploid males and diploid females. Although such haplodiploid species play key ecological roles and are threatened by environmental changes, no general framework exists for simulating their genetic evolution. Here, we use the SLiM simulation environment to build a novel model for individual-based forward simulation of genetic evolution in haplodiploids. We compare the fates of adaptive and deleterious mutations and find that selection on recessive mutations is more effective in haplodiploids than in diploids. Our open-source model will foster an understanding of the evolution of sociality and how ecologically important haplodiploid species may respond to changing environments.
Genome Biology and Evolution, Volume 14, Issue 5, May 2022, evac062 -
The era of reference genomes in conservation genomics
[DOI]
European Reference Genome Atlas (ERGA) Consortium
Progress in genome sequencing now enables the large-scale generation of reference genomes. Various international initiatives aim to generate reference genomes representing global biodiversity. These genomes provide unique insights into genomic diversity and architecture, thereby enabling comprehensive analyses of population and functional genomics, and are expected to revolutionize conservation genomics.
Trends in Ecology & Evolution, Volume 37, Issue 3, March 2022, Pages 197-202 -
Recurring adaptive introgression of a supergene variant that determines social organization
[DOI]
[Press release]
E Stolle*, R Pracana*, F López-Osorio, M K Priebe, GL Hernández, C Castillo-Carrillo, MC Arias, CI Paris, M Bollazzi, A Priyam, Y Wurm°
Introgression has been proposed as an essential source of adaptive genetic variation. However, a key barrier to adaptive introgression is that recombination can break down combinations of alleles that underpin many traits. This barrier might be overcome in supergene regions, where suppressed recombination leads to joint inheritance across many loci. Here, we study the evolution of a large supergene region that determines a major social and ecological trait in Solenopsis fire ants: whether colonies have one queen or multiple queens. Using coalescent-based phylogenies built from the genomes of 365 haploid fire ant males, we show that the supergene variant responsible for multiple-queen colonies evolved in one species and repeatedly spread to other species through introgressive hybridization. This finding highlights how supergene architecture can enable a complex adaptive phenotype to recurrently permeate species boundaries.
Nat Communications 13, 1180 (2022) -
Larger, more connected societies of ants have a higher prevalence of viruses
[DOI]
A Brahma, R Leon, GL Hernández, Y Wurm°
The benefits of cooperative living for foraging, nesting, defence and buffering environmental challenges lead animals with the most highly social lifestyles to dominate many ecosystems. However, living in larger, more highly connected groups should also increase the risks of pathogen exposure and transmission. While over long timescales selective responses could buffer the impacts of potential higher pathogen prevalence, similar processes are unlikely over short timescales. The red fire ant Solenopsis invicta is ideal for measuring the effects of group size on pathogen prevalence because two types of society coexist in this species: smaller single-nest single-queen colonies that are highly aggressive to their neighbours and larger multiple-queen colonies that exchange resources with neighbouring nests. We compare the presence of viruses between these two colony types using metagenomic sequence classification of RNA-sequencing reads. We find that queens from multiple-queen colonies have 8.3-times higher viral load and 1.5-times higher viral diversity than queens from single-queen colonies. This finding characterizes a rarely considered cost of transitions to more highly social living. Furthermore, our results show that highly social invertebrates can harbour many viruses.
Molecular Ecology (2022) 31:859-865 -
Genomic signatures of recent adaptation in a wild bumblebee
[DOI]
TJ Colgan, AN Arce, RJ Gill, A Ramos Rodrigues, A Kanteh, EJ Duncan, L Li, L Chittka, Y Wurm°
Environmental changes threaten insect pollinators, creating risks for agriculture and ecosystem stability. Despite their importance, we know little about how wild insects respond to environmental pressures. To understand the genomic bases of adaptation in an ecologically important pollinator, we analyzed genomes of Bombus terrestris bumblebees collected across Great Britain. We reveal extensive genetic diversity within this population, and strong signatures of recent adaptation throughout the genome affecting key processes including neurobiology and wing development. We also discover unusual features of the genome, including a region containing 53 genes that lacks genetic diversity in many bee species, and a horizontal gene transfer from a Wolbachia bacteria. Overall, the genetic diversity we observe and how it is distributed throughout the genome and the population should support the resilience of this important pollinator species to ongoing and future selective pressures. Applying our approach to more species should help understand how they can differ in their adaptive potential, and to develop conservation strategies for those most at risk.
Molecular Biology and Evolution, Volume 39, Issue 2, February 2022, msab366 -
Functional genomics of supergene-controlled behavior in the white-throated sparrow
[DOI]
PWH Holland, CD Jiggins, M Liedvogel, G Warren, Y Wurm
Supergenes are regions of suppressed recombination that may span hundreds of genes and can control variation in key ecological phenotypes. Since genetic analysis is made impossible by the absence of recombination between genes, it has been difficult to establish how individual genes within these regions contribute to supergene-controlled phenotypes. The white-throated sparrow is a classic example in which a supergene controls behavioral differences as well as distinct coloration that determines mate choice. A landmark study now demonstrates that differences between supergene variants in the promoter sequences of a hormone receptor gene change its expression and control changes in behavior. To unambiguously establish the link between genotype and phenotype, the authors used antisense oligonucleotides to alter the level of gene expression in a focal brain region targeted through a cannula. The study showcases a powerful approach to the functional genomic manipulation of a wild vertebrate species.
Faculty Reviews, Volume 10, 2021 -
Social isolation and group size are associated with divergent gene expression in the brain of ant queens
[DOI]
F Manfredini, C Martinez-Ruiz, Y Wurm, DW Shoemaker, MJF Brown
Social life and isolation pose a complex suite of challenges to organisms prompting significant changes in neural state. However, plasticity in how brains respond to social challenges remains largely unexplored. The fire ants Solenopsis invicta provide an ideal scenario for examining this. Fire ant queens may found colonies individually or in groups of up to 30 queens, depending on key factors such as density of newly mated queens and availability of nesting sites. We artificially manipulated availability of nesting sites to test how the brain responds to social vs. solitary colony founding at two key timepoints (early vs. late colony founding) and to group size (large vs. small groups). We adopted a powerful neurogenomic approach to identify even subtle differences of gene expression between treatment groups, and we built a global gene co‐expression network of the fire ant brain to identify gene modules specifically
Genes, Brain and Behavior. (2021)e12758 -
Parameter exploration improves the accuracy of long-read genome assembly
[DOI]
A Priyam, A Witwicka, A Brahma, E Stolle, Y Wurm°
Long-molecule sequencing is now routinely applied to generate high-quality reference genome assemblies. However, datasets differ in repeat composition, heterozygosity, read lengths and error profiles. The assembly parameters that provide the best results could thus differ across datasets. By integrating four complementary and biologically meaningful metrics, we show that simple fine-tuning of assembly parameters can substantially improve the quality of long-read genome assemblies. In particular, modifying estimates of sequencing error rates improves some metrics more than two-fold. We provide a flexible software, CompareGenomeQualities, that automates comparisons of assembly qualities for researchers wanting a straightforward mechanism for choosing among multiple assemblies.
bioRxiv 2021.05.28.446135 -
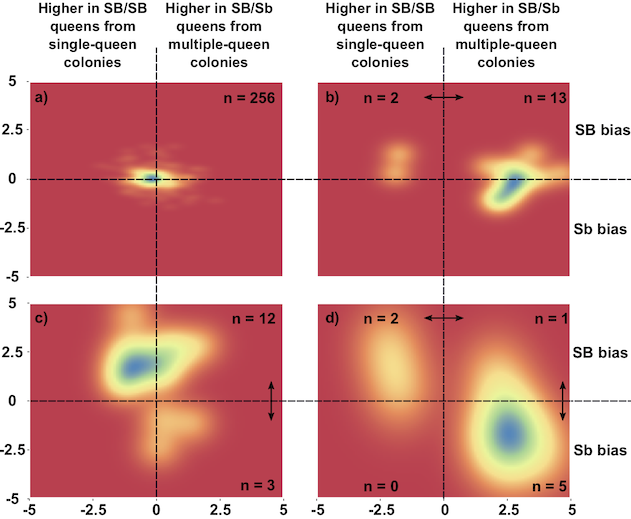 Genomic architecture and evolutionary antagonism drive allelic expression bias in the social supergene of red fire ants
[DOI]
C Martinez-Ruiz, R Pracana, E Stolle, CI Paris, RA Nichols, Y Wurm°
Genomic architecture and evolutionary antagonism drive allelic expression bias in the social supergene of red fire ants
[DOI]
C Martinez-Ruiz, R Pracana, E Stolle, CI Paris, RA Nichols, Y Wurm°Supergene regions maintain alleles of multiple genes in tight linkage through suppressed recombination. Despite their importance in determining complex phenotypes, our empirical understanding of early supergene evolution is limited. Here we focus on the young "social" supergene of fire ants, a powerful system for disentangling the effects of evolutionary antagonism and suppressed recombination. We hypothesize that gene degeneration and social antagonism shaped the evolution of the fire ant supergene, resulting in distinct patterns of gene expression. We test these ideas by identifying allelic differences between supergene variants, characterizing allelic expression across populations, castes and body parts, and contrasting allelic expression biases with differences in expression between social forms. We find strong signatures of gene degeneration and gene-specific dosage compensation. On this background, a small portion of the genes has the signature of adaptive responses to evolutionary antagonism between social forms.
eLife (2020) 9:e55862 -
Evolution: The legacy of endosymbiosis in ants
[DOI]
R Jackson, L Henry, Y Wurm
A symbiotic partnership with Blochmannia bacteria is thought to underpin the ecological success of carpenter ants. Disentangling the molecular interactions between the mutualistic partners supports an old hypothesis that many other ants also had similar symbioses and lost them.
Current Biology (2020) R1385 -
 Healthy pollinators: evaluating pesticides with molecular medicine approaches
[DOI]
[PDF]
F López-Osorio, Y Wurm°
Healthy pollinators: evaluating pesticides with molecular medicine approaches
[DOI]
[PDF]
F López-Osorio, Y Wurm°Pollinators have been declining worldwide, and pesticides have contributed to these declines. High-resolution approaches from molecular medicine can provide unparalleled insight into organismal physiology and health. Applying these approaches to pollinators can significantly improve the efficiency and sensitivity of pesticide research and evaluation, and thus the sustainability of modern agriculture.
Trends in Ecology & Evolution (2020) 35:380-3 -
Only a single taxonomically restricted gene family in the Drosophila melanogaster subgroup can be identified with high confidence
[DOI]
K Zile°, C Dessimoz, Y Wurm, J Masel
Taxonomically restricted genes (TRGs) are genes that are present only in one clade. Protein-coding TRGs may evolve de novo from previously non-coding sequences: functional ncRNA, introns or alternative reading frames of older protein-coding genes, or intergenic sequences. A major challenge in studying de novo genes is the need to avoid both false positives (non-functional open reading frames and/or functional genes that did not arise de novo) and false negatives. Here we search conservatively for high confidence TRGs as the most promising candidates for experimental studies, ensuring functionality through conservation across at least two species, and ensuring de novo status through examination of homologous non-coding sequences. Our pipeline also avoids ascertainment biases associated with preconceptions of how de novo genes are born. We identify one TRG family that evolved de novo in the Drosophila melanogaster subgroup. This TRG family contains single copy genes in D. simulans and D. sechellia. It originated in an intron of a well-established gene, sharing that intron with another well-established gene upstream. These TRGs contain an intron that pre-dates their ORF. These genes have not been previously reported as de novo originated, and to our knowledge they are the best Drosophila candidates identified so far for experimental studies aimed at elucidating the properties of de novo genes.
Genome Biology and Evolution (2020) evaa127 -
 Sequenceserver: a modern graphical user interface for custom BLAST databases
[DOI]
[Website]
[PDF]
[Supplementary]
A Priyam, B Woodcroft, V Rai, I Moghul, A Munagala, F Ter, H Chowdhary, I Pieniak, L Maynard, M Gibbins, H Moon, A Davis-Richardson, M Uludag, N Watson-Haigh, R Challis, H Nakamura, E Favreau, E Gómez, T Pluskal, G Leonard, W Rumpf, Y Wurm°
Sequenceserver: a modern graphical user interface for custom BLAST databases
[DOI]
[Website]
[PDF]
[Supplementary]
A Priyam, B Woodcroft, V Rai, I Moghul, A Munagala, F Ter, H Chowdhary, I Pieniak, L Maynard, M Gibbins, H Moon, A Davis-Richardson, M Uludag, N Watson-Haigh, R Challis, H Nakamura, E Favreau, E Gómez, T Pluskal, G Leonard, W Rumpf, Y Wurm°Comparing newly obtained and previously known nucleotide and amino-acid sequences underpins modern biological research. BLAST is a well-established tool for such comparisons but is challenging to use on new data sets. We combined a user-centric design philosophy with sustainable software development approaches to create Sequenceserver, a tool for running BLAST and visually inspecting BLAST results for biological interpretation. Sequenceserver uses simple algorithms to prevent potential analysis errors and provides flexible text-based and visual outputs to support researcher productivity. Our software can be rapidly installed for use by individuals or on shared servers.
Molecular Biology and Evolution (2019) msz185 -
Choosing the Best Gene Predictions with GeneValidator
[PDF]
I Moghul°, A Priyam, Y Wurm°
GeneValidator is a tool for determining whether the characteristics of newly predicted protein-coding genes are consistent with those of similar sequences in public databases. For this, it runs up to seven comparisons per gene. Results are shown in an HTML report containing summary statistics and graphical visualizations that aim to be useful for curators. Results are also presented in CSV and JSON formats for automated follow-up analysis. Here, we describe common usage scenarios of GeneValidator that use the JSON output results together with standard UNIX tools. We demonstrate how GeneValidator’s textual output can be used to filter and subset large gene sets effectively. First, we explain how low-scoring gene models can be identified and extracted for manual curation—for example, as input for genome browsers or gene annotation tools. Second, we show how GeneValidator’s HTML report can be regenerated from a filtered subset of GeneValidator’s JSON output. Subsequently, we demonstrate how GeneValidator’s GUI can be used to complement manual curation efforts. Additionally, we explain how GeneValidator can be used to merge information from multiple annotations by automatically selecting the higher-scoring gene model at each common gene locus. Finally, we show how GeneValidator analyses can be optimized when using large BLAST databases.
Gene Prediction. Methods in Molecular Biology (2019) Volume 1962; Chapter 16 -
Caste- and pesticide-specific effects of neonicotinoid pesticide exposure on gene expression in bumblebees.
[DOI]
TJ Colgan*, IK Fletcher*, AN Arce*, RJ Gill, A Ramos Rodrigues, E Stolle, L Chittka, Y Wurm°
Social bees are important insect pollinators of wildflowers and agricultural crops, making their reported declines a global concern. A major factor implicated in these declines is the widespread use of neonicotinoid pesticides. Indeed, recent research has demonstrated that exposure to low doses of these neurotoxic pesticides impairs bee behaviours important for colony function and survival. However, our understanding of the molecular‐genetic pathways that lead to such effects is limited, as is our knowledge of how effects may differ between colony members. To understand what genes and pathways are affected by exposure of bumblebee workers and queens to neonicotinoid pesticides, we implemented a transcriptome‐wide gene expression study. We chronically exposed Bombus terrestris colonies to either clothianidin or imidacloprid at field‐realistic concentrations while controlling for factors including colony social environment and worker age. We reveal that genes involved in important biological processes including mitochondrial function are differentially expressed in response to neonicotinoid exposure. Additionally, clothianidin exposure had stronger effects on gene expression amplitude and alternative splicing than imidacloprid. Finally, exposure affected workers more strongly than queens. Our work demonstrates how RNA‐Seq transcriptome profiling can provide detailed novel insight on the mechanisms mediating pesticide toxicity to a key insect pollinator.
Molecular Ecology (2019) 28: 1964–74 -
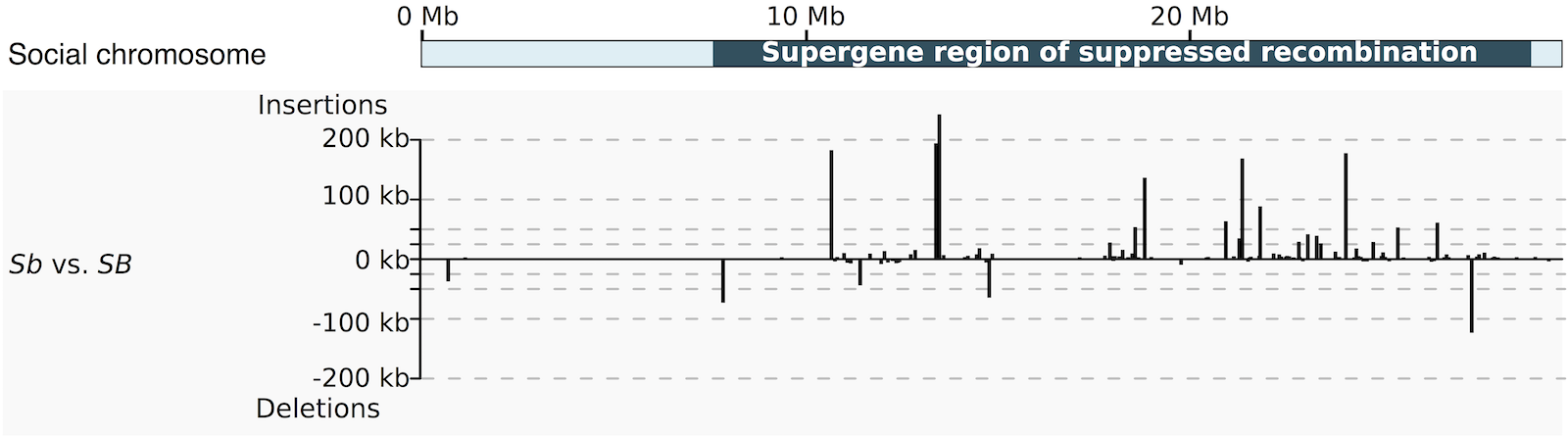 Degenerative expansion of a young supergene
[DOI]
[PDF]
[Supplementary]
E Stolle°, R Pracana, P Howard, CI Paris, SJ Brown, CA Castillo-Carrillo, SJ Rossiter, Y Wurm°
Degenerative expansion of a young supergene
[DOI]
[PDF]
[Supplementary]
E Stolle°, R Pracana, P Howard, CI Paris, SJ Brown, CA Castillo-Carrillo, SJ Rossiter, Y Wurm°Long-term suppression of recombination ultimately leads to gene loss, as demonstrated by the depauperate Y and W chromosomes of long-established pairs of XY and ZW chromosomes. The young social supergene of the Solenopsis invicta red fire ant provides a powerful system to examine the effects of suppressed recombination over a shorter timescale. The two variants of this supergene are carried by a pair of heteromorphic chromosomes, referred to as the social B and social b (SB and Sb) chromosomes. The Sb variant of this supergene changes colony social organization and has an inheritance pattern similar to a Y or W chromosome because it is unable to recombine. We used high-resolution optical mapping, k-mer distribution analysis, and quantification of repetitive elements on haploid ants carrying alternate variants of this young supergene region.
We find that instead of shrinking, the Sb variant of the supergene has increased in length by more than 30%. Surprisingly, only a portion of this length increase is due to consistent increases in the frequency of particular classes of repetitive elements. Instead, haplotypes of this supergene variant differ dramatically in the amounts of other repetitive elements, indicating that the accumulation of repetitive elements is a heterogeneous and dynamic process. This is the first comprehensive demonstration of degenerative expansion in an animal and shows that it occurs through nonlinear processes during the early evolution of a region of suppressed recombination.
Molecular Biology and Evolution (2019) -
The first draft genomes of the ant Formica exsecta, and its Wolbachia endosymbiont reveal extensive gene transfer from endosymbiont to host.
[DOI]
K Dhaygude, A Nair, H Johansson, Y Wurm, L Sundström
Adapting to changes in the environment is the foundation of species survival, and is usually thought to be a gradual process. However, transposable elements (TEs), epigenetic modifications, and/or genetic material acquired from other organisms by means of horizontal gene transfer (HGTs), can also lead to novel adaptive traits. Social insects form dense societies, which attract and maintain extra- and intracellular accessory inhabitants, which may facilitate gene transfer between species. The wood ant Formica exsecta (Formicidae; Hymenoptera), is a common ant species throughout the Palearctic region. The species is a well-established model for studies of ecological characteristics and evolutionary conflict. In this study, we sequenced and assembled draft genomes for F. exsecta and its endosymbiont Wolbachia. The F. exsecta draft genome is 277.7 Mb long; we identify 13,767 protein coding genes, for which we provide gene ontology and protein domain annotations. This is also the first report of a Wolbachia genome from ants, and provides insights into the phylogenetic position of this endosymbiont. We also identified multiple horizontal gene transfer events (HGTs) from Wolbachia to F. exsecta. Some of these HGTs have also occurred in parallel in multiple other insect genomes, highlighting the extent of HGTs in eukaryotes. We present the first draft genome of ant F. exsecta, and its endosymbiont Wolbachia (wFex), and show considerable rates of gene transfer from the symbiont to the host. We expect that especially the F. exsecta genome will be valuable resource in further exploration of the molecular basis of the evolution of social organization.
BMC Genomics (2019) 20:301 -
 Foraging bumblebees acquire a preference for neonicotinoid-treated food with prolonged exposure
[DOI]
AN Arce, A Ramos Rodrigues, J Yu, TJ Colgan, Y Wurm, RJ Gill
Foraging bumblebees acquire a preference for neonicotinoid-treated food with prolonged exposure
[DOI]
AN Arce, A Ramos Rodrigues, J Yu, TJ Colgan, Y Wurm, RJ GillSocial bees represent an important group of pollinating insects that can be exposed to potentially harmful pesticides when foraging on treated or contaminated flowering plants. To investigate if such exposure is detrimental to bees, many studies have exclusively fed individuals with pesticide-spiked food, informing us about the hazard but not necessarily the risk of exposure. While such studies are important to establish the physiological and behavioural effects on individuals, they do not consider the possibility that the risk of exposure may change over time. For example, many pesticide assays exclude potential behavioural adaptations to novel toxins, such as rejection of harmful compounds by choosing to feed on an uncontaminated food source, thus behaviourally lowering the risk of exposure. In this paper, we conducted an experiment over 10 days in which bumblebees could forage on an array of sucrose feeders containing 0, 2 and 11 parts per billion of the neonicotinoid pesticide thiamethoxam. This more closely mimics pesticide exposure in the wild by allowing foraging bees to (i) experience a field realistic range of pesticide concentrations across a chronic exposure period, (ii) have repeated interactions with the pesticide in their environment, and (iii) retain the social cues associated with foraging by using whole colonies. We found that the proportion of visits to pesticide-laced feeders increased over time, resulting in greater consumption of pesticide-laced sucrose relative to untreated sucrose. After changing the spatial position of each feeder, foragers continued to preferentially visit the pesticide-laced feeders which indicates that workers can detect thiamethoxam and alter their behaviour to continue feeding on it. The increasing preference for consuming the neonicotinoid-treated food therefore increases the risk of exposure for the colony during prolonged pesticide exposure. Our results highlight the need to incorporate attractiveness of pesticides to foraging bees (and potentially other insect pollinators) in addition to simply considering the proportion of pesticide-contaminated floral resources within the foraging landscape.
Proceedings of the Royal Society B: Biological Sciences (2018) 285:20180655 -
Genes and genomic processes underpinning the social lives of ants
[DOI]
[PDF]
E Favreau*, C Martínez-Ruiz*, L Santiago Rodrigues*, R L Hammond°, Y Wurm°
The >15 000 ant species are all highly social and show great variation in colony organization, complexity and behavior. The mechanisms by which such sociality evolved, as well as those underpinning the elaboration of ant societies since their ∼140 million year old common ancestor, have long been pondered. Here, we review recent insights generated using various genomic approaches. This includes understanding the molecular mechanisms underlying caste differentiation and the diversity of social structures, studying the impact of eusociality on genomic evolutionary rates, and investigating gene expression changes associated with differences in lifespan between castes. Furthermore, functional studies involving RNAi and CRISPR have recently been successfully applied to ants, opening the door to exciting research that promises to revolutionize the understanding of the evolution and diversification of social living.
Current Opinion in Insect Science (2018), 25:83-90 -
The Global Ant Genomics Alliance (GAGA)
[PDF]
The Global Ant Genomics Alliance (GAGA) Consortium
Myrmecological News, Volume 25, October 2017, Pages 61-66, Vienna -
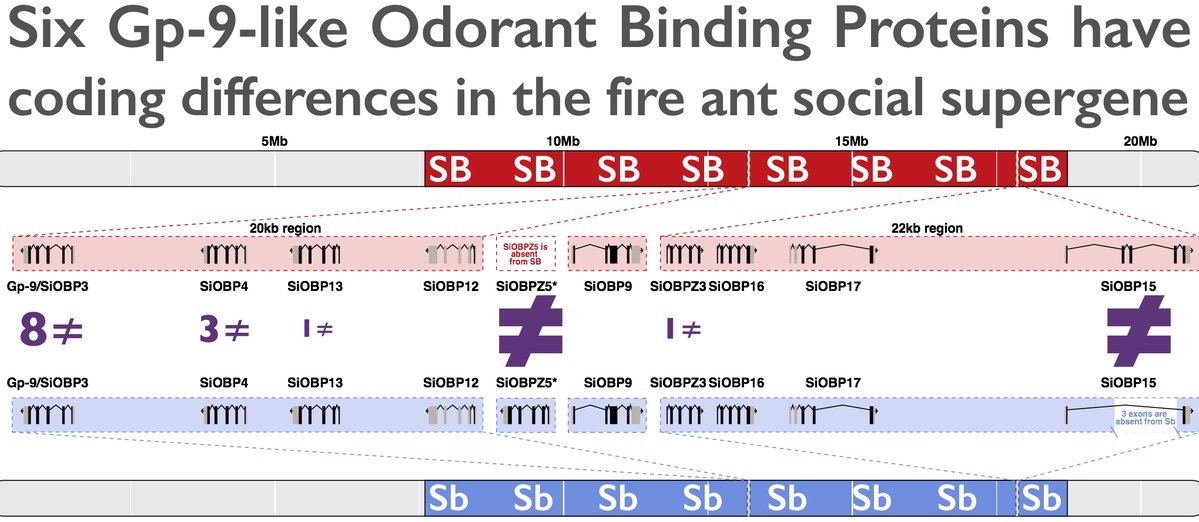 Fire ant social chromosomes: Differences in number, sequence and expression of odorant binding proteins
[DOI]
[PDF]
R Pracana, I Levantis, C Martínez-Ruiz, E Stolle, A Priyam, Y Wurm°
Fire ant social chromosomes: Differences in number, sequence and expression of odorant binding proteins
[DOI]
[PDF]
R Pracana, I Levantis, C Martínez-Ruiz, E Stolle, A Priyam, Y Wurm°Variation in social behavior is common yet our knowledge of the mechanisms underpinning its evolution is limited. The fire ant Solenopsis invicta provides a textbook example of a Mendelian element controlling social organization: alternate alleles of a genetic element first identified as encoding an odorant binding protein (OBP) named Gp‐9 determine whether a colony accepts one or multiple queens. The potential roles of such a protein in perceiving olfactory cues and evidence of positive selection on its amino acid sequence made it an appealing candidate gene. However, we recently showed that recombination is suppressed between Gp‐9 and hundreds of other genes as part of a >19 Mb supergene‐like region carried by a pair of social chromosomes. This finding raises the need to reassess the potential role of Gp‐9 . We identify 23 OBPs in the fire ant genome assembly, including nine located in the region of suppressed recombination with Gp‐9 . For six of these, the alleles carried by the two variants of the supergene‐like region differ in protein‐coding sequence and thus likely in function, with Gp‐9 showing the strongest evidence of positive selection. We identify an additional OBP specific to the Sb variant of the region. Finally, we find that 14 OBPs are differentially expressed between single‐ and multiple‐queen colonies. These results are consistent with multiple OBPs playing a role in determining social structure.
Evolution Letters (2017) 1:199-210 -
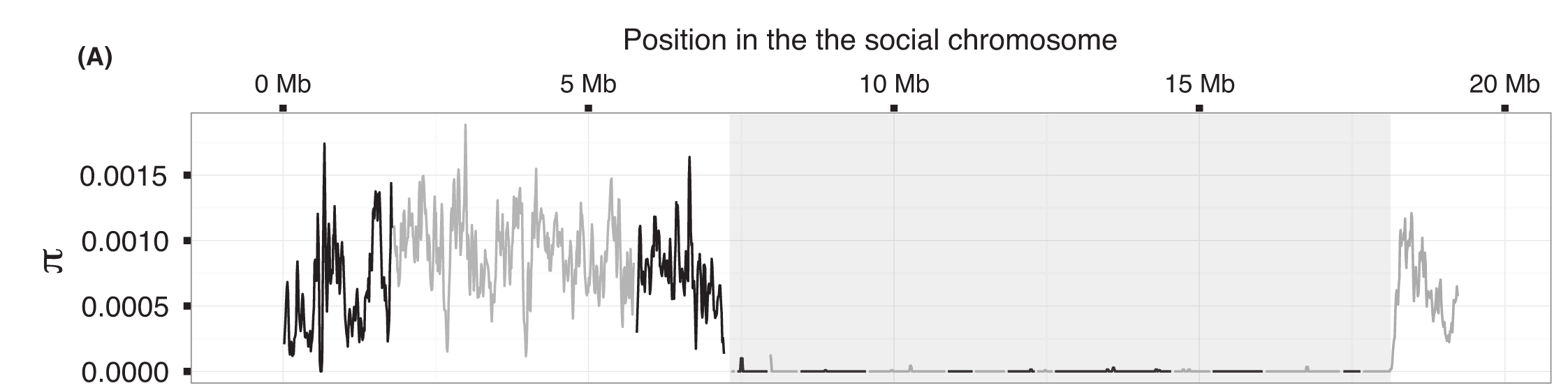 The fire ant social chromosome supergene variant Sb shows low diversity but high divergence from SB
[DOI]
[PDF]
R Pracana, A Priyam, I Levantis, R Nichols, Y Wurm°
The fire ant social chromosome supergene variant Sb shows low diversity but high divergence from SB
[DOI]
[PDF]
R Pracana, A Priyam, I Levantis, R Nichols, Y Wurm°Variation in social behaviour is common, yet little is known about the genetic architectures underpinning its evolution. A rare exception is in the fire ant Solenopsis invicta : Alternative variants of a supergene region determine whether a colony will have exactly one or up to dozens of queens. The two variants of this region are carried by a pair of ‘social chromosomes’, SB and Sb, which resemble a pair of sex chromosomes. Recombination is suppressed between the two chromosomes in the supergene region. While the X‐like SB can recombine with itself in SB/SB queens, recombination is effectively absent in the Y‐like Sb because Sb/Sb queens die before reproducing. Here, we analyse whole‐genome sequences of eight haploid SB males and eight haploid Sb males. We find extensive SB–Sb differentiation throughout the >19‐Mb‐long supergene region. We find no evidence of ‘evolutionary strata’ with different levels of divergence comparable to those reported in several sex chromosomes. A high proportion of substitutions between the SB and Sb haplotypes are nonsynonymous, suggesting inefficacy of purifying selection in Sb sequences, similar to that for Y‐linked sequences in XY systems. Finally, we show that the Sb haplotype of the supergene region has 635‐fold less nucleotide diversity than the rest of the genome. We discuss how this reduction could be due to a recent selective sweep affecting Sb specifically or associated with a population bottleneck during the invasion of North America by the sampled population.
Molecular Ecology (2017) 26:2864-79 -
Impact of controlled neonicotinoid exposure on bumblebees in a realistic field setting
[DOI]
A Arce, T David, E Randall, A Ramos Rodrigues, T Colgan, Y Wurm, R Gill
Pesticide exposure has been implicated as a contributor to insect pollinator declines. In social bees, which are crucial pollination service providers, the effect of low‐level chronic exposure is typically non‐lethal leading researchers to consider whether exposure induces sublethal effects on behaviour and whether such impairment can affect colony development. Studies under laboratory conditions can control levels of pesticide exposure and elucidate causative effects, but are often criticized for being unrealistic. In contrast, field studies can monitor bee responses under a more realistic pesticide exposure landscape; yet typically such findings are limited to correlative results and can lack true controls or sufficient replication. We attempt to bridge this gap by exposing bumblebees to known amounts of pesticides when colonies are placed in the field. Using 20 bumblebee colonies, we assess the consequences of exposure to the neonicotinoid clothianidin, provided in sucrose at a concentration of five parts per billion, over 5 weeks. We monitored foraging patterns and pollen collecting performance from 3282 bouts using either a non‐invasive photographic assessment, or by extracting the pollen from returning foragers. We also conducted a full colony census at the beginning and end of the experiment. In contrast to studies on other neonicotinoids, showing clear impairment to foraging behaviours, we detected only subtle changes to patterns of foraging activity and pollen foraging during the course of the experiment. However, our colony census measures showed a more pronounced effect of exposure, with fewer adult workers and sexuals in treated colonies after 5 weeks. Synthesis and applications . Pesticide‐induced impairments on colony development and foraging could impact on the pollination service that bees provide. Therefore, our findings, that bees show subtle changes in foraging behaviour and reductions in colony size after exposure to a common pesticide, have important implications and help to inform the debate over whether the benefits of systemic pesticide application to flowering crops outweigh the costs. We propose that our methodology is an important advance to previous semi‐field methods and should be considered when considering improvements to current ecotoxicological guidelines for pesticide risk assessment.
Journal of Applied Ecology (2016) 54:1199–1208 -
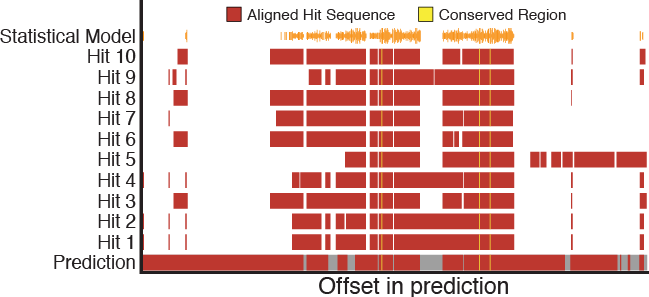 GeneValidator: identify problems with protein-coding gene predictions
[DOI]
[Website]
[PDF]
M Drăgan*, I Moghul*, A Priyam, C Bustos, Y Wurm°
GeneValidator: identify problems with protein-coding gene predictions
[DOI]
[Website]
[PDF]
M Drăgan*, I Moghul*, A Priyam, C Bustos, Y Wurm°Genomes of emerging model organisms are now being sequenced at very low cost. However, obtaining accurate gene predictions remains challenging: even the best gene prediction algorithms make substantial errors and can jeopardize subsequent analyses. Therefore, many predicted genes must be time-consumingly visually inspected and manually curated. We developed GeneValidator (GV) to automatically identify problematic gene predictions and to aid manual curation. For each gene, GV performs multiple analyses based on comparisons to gene sequences from large databases. The resulting report identifies problematic gene predictions and includes extensive statistics and graphs for each prediction to guide manual curation efforts. GV thus accelerates and enhances the work of biocurators and researchers who need accurate gene predictions from newly sequenced genomes.
Bioinformatics (2016) 32:1559-61 -
Transcriptomic identification of starfish neuropeptide precursors yields new insights into neuropeptide evolution
[DOI]
[PDF]
D Semmens, O Mirabeau, I Moghul, M Pancholi, Y Wurm, M Elphick
Neuropeptides are evolutionarily ancient mediators of neuronal signalling in nervous systems. With recent advances in genomics/transcriptomics, an increasingly wide range of species has become accessible for molecular analysis. The deuterostomian invertebrates are of particular interest in this regard because they occupy an ‘intermediate' position in animal phylogeny, bridging the gap between the well-studied model protostomian invertebrates (e.g. Drosophila melanogaster, Caenorhabditis elegans) and the vertebrates. Here we have identified 40 neuropeptide precursors in the starfish Asterias rubens, a deuterostomian invertebrate from the phylum Echinodermata. Importantly, these include kisspeptin-type and melanin-concentrating hormone-type precursors, which are the first to be discovered in a non-chordate species. Starfish tachykinin-type, somatostatin-type, pigment-dispersing factor-type and corticotropin-releasing hormone-type precursors are the first to be discovered in the echinoderm/ambulacrarian clade of the animal kingdom. Other precursors identified include vasopressin/oxytocin-type, gonadotropin-releasing hormone-type, thyrotropin-releasing hormone-type, calcitonin-type, cholecystokinin/gastrin-type, orexin-type, luqin-type, pedal peptide/orcokinin-type, glycoprotein hormone-type, bursicon-type, relaxin-type and insulin-like growth factor-type precursors. This is the most comprehensive identification of neuropeptide precursor proteins in an echinoderm to date, yielding new insights into the evolution of neuropeptide signalling systems. Furthermore, these data provide a basis for experimental analysis of neuropeptide function in the unique context of the decentralized, pentaradial echinoderm bauplan.
Open Biology (2016) 6:150224 -
 Arthropod genomics beyond fruit flies: bridging the gap between proximate and ultimate causation
[DOI]
Arthropod genomics beyond fruit flies: bridging the gap between proximate and ultimate causation
[DOI]
Y Wurm
Briefings in Functional Genomics (2015) 14:381-3 -
Ant genomics (Hymenoptera: Formicidae): challenges to overcome and opportunities to seize
[PDF]
Y Wurm°*, S Nygaard°*
Myrmecological News (2015) 21:59-72 -
Desktop as a service supporting environmental'omics
[PDF]
DCH Wallom, T Booth, A Bowery, B Collier, P Kershaw, A Priyam, Y Wurm, D Field
Within the Environmental 'omics community Bio-Linux is a widely used tool. This has the advantage of providing in a single deliverable package all necessary software and tools to support common analyses. With the growth in data volumes within the community and increasing constraints on user access and control over their own desktops an alternative delivery method of Bio-Linux and, in future, the Docker container environment is necessary. Within the EOS Cloud project we have constructed a Desktop as a Service system to centrally host virtual machines with these tools preconfigured and maintained. To enable efficient use of the resources we have enabled user controlled resource scaling so that users are able to utilise small scale VMs for task configuration and data manipulation and boost to a larger scale to run analysis applications all the while maintaining the user environment in a consistent manner. Alongside this within the project we have been developed tools to simplify the increasingly popular Docker software usage model. This includes ensure uniformity of behaviour between the host system and the running Docker container. Within the invitation only trial user community we identify two different exemplars groups and explain their usage and how the products and services developed within the project are useful for them. We conclude discussing the useful nature of Desktop as a Service, how it is of great benefit to the bioinformatics community but could also be of great use elsewhere, where the need for a stable user environment with applications already available that do not rely on local ICT support.
IEEE 11th International Conference on e-Science (2015) 214 -
Avoid having to retract your genomics analysis
[DOI]
[PDF]
Y Wurm
The dramatic plunge in DNA sequencing costs means that a single MSc or PhD student can now generate data that would have cost $15,000,000 only ten years ago. We are thus leaping from lab-notebook-scale science to research that requires extensive programming, statistics and high performance computing. This is exciting & empowering - in particular for small teams working on emerging model organisms that lacked genomic resources. But with great powers come great responsibilities... and risks of doing things wrong. These risks are far greater for genome biologists than, say physicists or astronomers who have strong traditions of working with large datasets.
The Winnower (2015) 2:e143696.68941 -
Social chromosome variants differentially affect queen determination and the survival of workers in the fire ant Solenopsis invicta
[DOI]
[PDF]
SD Buechel*, Y Wurm*, L Keller
Intraspecific variation in social organization is common, yet the underlying causes are rarely known. An exception is the fire ant S olenopsis invicta in which the existence of two distinct forms of social colony organization is under the control of the two variants of a pair of social chromosomes, SB and S b. Colonies containing exclusively SB /SB workers accept only one single queen and she must be SB /SB . By contrast, when colonies contain more than 10% of SB /Sb workers, they accept several queens but only SB /Sb queens. The variants of the social chromosome are associated with several additional important phenotypic differences, including the size, fecundity and dispersal strategies of queens, aggressiveness of workers, and sperm count in males. However, little is known about whether social chromosome variants affect fitness in other life stages. Here, we perform experiments to determine whether differential selection occurs during development and in adult workers. We find evidence that the S b variant of the social chromosome increases the likelihood of female brood to develop into queens and that adult SB /S b workers, the workers that cull SB /SB queens, are overrepresented in comparison to SB /SB workers. This demonstrates that supergenes such as the social chromosome can have complex effects on phenotypes at various stages of development.
Molecular Ecology (2014) 23:5117-27 -
Transposable element islands facilitate adaptation to novel environments in an invasive species
[DOI]
[PDF]
L Schrader, J Kim, D Ence, A Zimin, A Klein, K Wyschetzki, T Weichselgartner, C Kemena, J Stökl, E Schultner, Y Wurm, C Smith, M Yandell, J Heinze, J Gadau, J Oettler
Adaptation requires genetic variation, but founder populations are generally genetically depleted. Here we sequence two populations of an inbred ant that diverge in phenotype to determine how variability is generated. Cardiocondyla obscurior has the smallest of the sequenced ant genomes and its structure suggests a fundamental role of transposable elements (TEs) in adaptive evolution. Accumulations of TEs (TE islands) comprising 7.18% of the genome evolve faster than other regions with regard to single-nucleotide variants, gene/exon duplications and deletions and gene homology. A non-random distribution of gene families, larvae/adult specific gene expression and signs of differential methylation in TE islands indicate intragenomic differences in regulation, evolutionary rates and coalescent effective population size. Our study reveals a tripartite interplay between TEs, life history and adaptation in an invasive species.
Nature Communications (2014) 5:5495 -
Effects of ploidy and sex-locus genotype on gene expression patterns in the fire ant Solenopsis invicta
[DOI]
[PDF]
M Nipitwattanaphon, J Wang, K Ross, O Riba-Grognuz, Y Wurm, C Khurewathanakul, L Keller
Males in many animal species differ greatly from females in morphology, physiology and behaviour. Ants, bees and wasps have a haplodiploid mechanism of sex determination whereby unfertilized eggs become males while fertilized eggs become females. However, many species also have a low frequency of diploid males, which are thought to develop from diploid eggs when individuals are homozygous at one or more sex determination loci. Diploid males are morphologically similar to haploids, though often larger and typically sterile. To determine how ploidy level and sex-locus genotype affect gene expression during development, we compared expression patterns between diploid males, haploid males and females (queens) at three developmental timepoints in Solenopsis invicta. In pupae, gene expression profiles of diploid males were very different from those of haploid males but nearly identical to those of queens. An unexpected shift in expression patterns emerged soon after adult eclosion, with diploid male patterns diverging from those of queens to resemble those of haploid males, a pattern retained in older adults. The finding that ploidy level effects on early gene expression override sex effects (including genes implicated in sperm production and pheromone production/perception) may explain diploid male sterility and lack of worker discrimination against them during development.
Proceedings of the Royal Society B: Biological Sciences (2014) 281:20141776 -
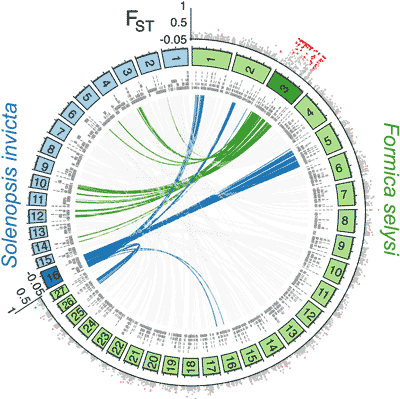 Convergent genetic architecture underlies social organization in ants
[DOI]
[PDF]
J Purcell, A Brelsford, Y Wurm, N Perrin, M Chapuisat
Convergent genetic architecture underlies social organization in ants
[DOI]
[PDF]
J Purcell, A Brelsford, Y Wurm, N Perrin, M ChapuisatComplex adaptive polymorphisms are common in nature, but what mechanisms maintain the underlying favorable allelic combinations? The convergent evolution of polymorphic social organization in two independent ant species provides a great opportunity to investigate how genomes evolved under parallel selection. Here, we demonstrate that a large, nonrecombining “social chromosome” is associated with social organization in the Alpine silver ant, Formica selysi. This social chromosome shares architectural characteristics with that of the fire ant Solenopsis invicta, but the two show no detectable similarity in gene content. The discovery of convergence at two levels—the phenotype and the genetic architecture associated with alternative social forms—points at general genetic mechanisms underlying transitions in social organization. More broadly, our findings are consistent with recent theoretical studies suggesting that suppression of recombination plays a key role in facilitating coordinated shifts in coadapted traits.
Current Biology (2014) 24:2728-32 -
Discovery and molecular characterization of an ambisense densovirus from South American populations of Solenopsis invicta
[DOI]
[PDF]
S Valles, D Shoemaker, Y Wurm, C Strong, L Varone, J Becnel, P Shirk
In an effort to discover viruses as classical biological control agents, a metatranscriptomics/pyrosequencing approach was used to survey native Solenopsis invicta collected exclusively in Argentina. A new virus was discovered with characteristics consistent with the family Parvoviridae, subfamily Densovirinae. The virus, tentatively named Solenopsis invicta densovirus (SiDNV), represents the first DNA virus discovered in ants (Formicidae) and the first densovirus in a hymenopteran insect. The ambisense genome was 5280 nucleotides in length and the termini possessed asymmetrically positioned inverted terminal repeats, formed hairpin loops, and had transcriptional regulatory elements including CAAT and TATA sites. Phylogenetic analysis revealed that SiDNV belongs to a group that includes two other densoviruses found in insects (Acheta domestica densovirus and Planococcus citri densovirus). SiDNV was prevalent in fire ants from Argentina but completely absent in fire ants found in the USA indicating that this virus has potential for biological control of introduced S. invicta.
Biological Control (2013) 67:431-39 -
Vitellogenin underwent subfunctionalization to acquire caste and behavioral specific expression in the harvester ant Pogonomyrmex barbatus
[DOI]
[PDF]
M Corona, J Zhang, R Libbrecht, Y Wurm, O Riba-Grognuz, R Studer, L Keller
The reproductive ground plan hypothesis (RGPH) proposes that the physiological pathways regulating reproduction were co-opted to regulate worker division of labor. Support for this hypothesis in honeybees is provided by studies demonstrating that the reproductive potential of workers, assessed by the levels of vitellogenin (Vg), is linked to task performance. Interestingly, contrary to honeybees that have a single Vg ortholog and potentially fertile nurses, the genome of the harvester ant Pogonomyrmex barbatus harbors two Vg genes (Pb_Vg1 and Pb_Vg2) and nurses produce infertile trophic eggs. P. barbatus, thus, provides a unique model to investigate whether Vg duplication in ants was followed by subfunctionalization to acquire reproductive and non-reproductive functions and whether Vg reproductive function was co-opted to regulate behavior in sterile workers. To investigate these questions, we compared the expression patterns of P. barbatus Vg genes and analyzed the phylogenetic relationships and molecular evolution of Vg genes in ants. qRT-PCRs revealed that Pb_Vg1 is more highly expressed in queens compared to workers and in nurses compared to foragers. By contrast, the level of expression of Pb_Vg2 was higher in foragers than in nurses and queens. Phylogenetic analyses show that a first duplication of the ancestral Vg gene occurred after the divergence between the poneroid and formicoid clades and subsequent duplications occurred in the lineages leading to Solenopsis invicta, Linepithema humile and Acromyrmex echinatior. The initial duplication resulted in two Vg gene subfamilies preferentially expressed in queens and nurses (subfamily A) or in foraging workers (subfamily B). Finally, molecular evolution analyses show that the subfamily A experienced positive selection, while the subfamily B showed overall relaxation of purifying selection. Our results suggest that in P. barbatus the Vg gene underwent subfunctionalization after duplication to acquire caste- and behavior- specific expression associated with reproductive and non-reproductive functions, supporting the validity of the RGPH in ants.
PLoS Genetics (2013) 9:e1003730 -
Sociogenomics of cooperation and conflict during colony founding in the fire ant Solenopsis invicta
[DOI]
[PDF]
F Manfredini, N Tsutsui, O Riba-Grognuz, Y Wurm, L Keller, D Shoemaker, C Grozinger
One of the fundamental questions in biology is how cooperative and altruistic behaviors evolved. The majority of studies seeking to identify the genes regulating these behaviors have been performed in systems where behavioral and physiological differences are relatively fixed, such as in the honey bee. During colony founding in the monogyne (one queen per colony) social form of the fire ant Solenopsis invicta, newly-mated queens may start new colonies either individually (haplometrosis) or in groups (pleometrosis). However, only one queen (the “winner”) in pleometrotic associations survives and takes the lead of the young colony while the others (the “losers”) are executed. Thus, colony founding in fire ants provides an excellent system in which to examine the genes underpinning cooperative behavior and how the social environment shapes the expression of these genes. We developed a new whole genome microarray platform for S. invicta to characterize the gene expression patterns associated with colony founding behavior. First, we compared haplometrotic queens, pleometrotic winners and pleometrotic losers. Second, we manipulated pleometrotic couples in order to switch or maintain the social ranks of the two cofoundresses. Haplometrotic and pleometrotic queens differed in the expression of genes involved in stress response, aging, immunity, reproduction and lipid biosynthesis. Smaller sets of genes were differentially expressed between winners and losers. In the second experiment, switching social rank had a much greater impact on gene expression patterns than the initial/final rank. Expression differences for several candidate genes involved in key biological processes were confirmed using qRT-PCR. Our findings indicate that, in S. invicta, social environment plays a major role in the determination of the patterns of gene expression, while the queen's physiological state is secondary. These results highlight the powerful influence of social environment on regulation of the genomic state, physiology and ultimately, social behavior of animals.
PLoS Genetics (2013) 9:e1003633 -
Comparative genomics of chemosensory protein genes reveals rapid evolution and positive selection in ant-specific duplicates
[DOI]
[PDF]
J Kulmuni°, Y Wurm, P Pamilo
Gene duplications can have a major role in adaptation, and gene families underlying chemosensation are particularly interesting due to their essential role in chemical recognition of mates, predators and food resources. Social insects add yet another dimension to the study of chemosensory genomics, as the key components of their social life rely on chemical communication. Still, chemosensory gene families are little studied in social insects. Here we annotated chemosensory protein (CSP) genes from seven ant genomes and studied their evolution. The number of functional CSP genes ranges from 11 to 21 depending on species, and the estimated rates of gene birth and death indicate high turnover of genes. Ant CSP genes include seven conservative orthologous groups present in all the ants, and a group of genes that has expanded independently in different ant lineages. Interestingly, the expanded group of genes has a differing mode of evolution from the orthologous groups. The expanded group shows rapid evolution as indicated by a high dN/dS (nonsynonymous to synonymous changes) ratio, several sites under positive selection and many pseudogenes, whereas the genes in the seven orthologous groups evolve slowly under purifying selection and include only one pseudogene. These results show that adaptive changes have played a role in ant CSP evolution. The expanded group of ant-specific genes is phylogenetically close to a conservative orthologous group CSP7, which includes genes known to be involved in ant nestmate recognition, raising an interesting possibility that the expanded CSPs function in ant chemical communication.
Heredity (2013) 110:538-47 -
 A Y-like social chromosome causes alternative colony organization in fire ants
[DOI]
[PDF]
J Wang*°, Y Wurm*°, M Nipitwattanaphon, O Riba-Grognuz, Y Huang, D Shoemaker, L Keller°
A Y-like social chromosome causes alternative colony organization in fire ants
[DOI]
[PDF]
J Wang*°, Y Wurm*°, M Nipitwattanaphon, O Riba-Grognuz, Y Huang, D Shoemaker, L Keller°Intraspecific variability in social organization is common, yet the underlying causes are rarely known. In the fire ant Solenopsis invicta, the existence of two divergent forms of social organization is under the control of a single Mendelian genomic element marked by two variants of an odorant-binding protein gene. Here we characterize the genomic region responsible for this important social polymorphism, and show that it is part of a pair of heteromorphic chromosomes that have many of the key properties of sex chromosomes. The two variants, hereafter referred to as the social B and social b (SB and Sb) chromosomes, are characterized by a large region of approximately 13 megabases (55% of the chromosome) in which recombination is completely suppressed between SB and Sb. Recombination seems to occur normally between the SB chromosomes but not between Sb chromosomes because Sb/Sb individuals are non-viable. Genomic comparisons revealed limited differentiation between SB and Sb, and the vast majority of the 616 genes identified in the non-recombining region are present in the two variants. The lack of recombination over more than half of the two heteromorphic social chromosomes can be explained by at least one large inversion of around 9 megabases, and this absence of recombination has led to the accumulation of deleterious mutations, including repetitive elements in the non-recombining region of Sb compared with the homologous region of SB. Importantly, most of the genes with demonstrated expression differences between individuals of the two social forms reside in the non-recombining region. These findings highlight how genomic rearrangements can maintain divergent adaptive social phenotypes involving many genes acting together by locally limiting recombination.
Nature (2013) 493:664-8 -
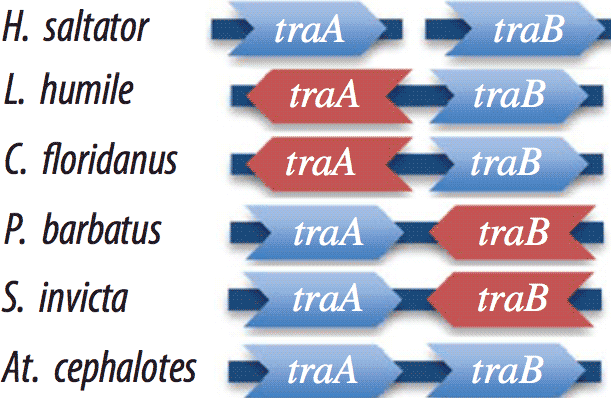 Duplication and concerted evolution in a master sex determiner under balancing selection
[DOI]
[PDF]
E Privman, Y Wurm°, L Keller°
Duplication and concerted evolution in a master sex determiner under balancing selection
[DOI]
[PDF]
E Privman, Y Wurm°, L Keller°The transformer (tra) gene is a key regulator in the signalling hierarchy controlling all aspects of somatic sexual differentiation in Drosophila and other insects. Here, we show that six of the seven sequenced ants have two copies of tra. Surprisingly, the two paralogues are always more similar within species than among species. Comparative sequence analyses indicate that this pattern is owing to the ongoing concerted evolution after an ancestral duplication rather than independent duplications in each of the six species. In particular, there was strong support for inter-locus recombination between the paralogues of the ant Atta cephalotes. In the five species where the location of paralogues is known, they are adjacent to each other in four cases and separated by only few genes in the fifth case. Because there have been extensive genomic rearrangements in these lineages, this suggests selection acting to conserve their synteny. In three species, we also find a signature of positive selection in one of the paralogues. In three bee species where information is available, the tra gene is also duplicated, the copies are adjacent and in at least one species there was recombination between paralogues. These results suggest that concerted evolution plays an adaptive role in the evolution of this gene family.
Proceedings of the Royal Society B: Biological Sciences (2013) 280:20122968 -
Social insect genomes exhibit dramatic evolution in gene composition and regulation while preserving regulatory features linked to sociality
[DOI]
[PDF]
D Simola, L Wissler, G Donahue, R Waterhouse, M Helmkampf, J Roux, S Nygaard, K Glastad, D Hagen, L Viljakainen, J Reese, B Hunt, D Graur, E Elhaik, E Kriventseva, J Wen, B Parker, E Cash, E Privman, C Childers, M Munoz-torres, J Boomsma, E Bornberg-bauer, C Currie, C Elsik, G Suen, M Goodisman, L Keller, J Liebig, A Rawls, D Reinberg, C Smith, C Smith, N Tsutsui, Y Wurm, E Zdobnov, S Berger, J Gadau
Genomes of eusocial insects code for dramatic examples of phenotypic plasticity and social organization. We compared the genomes of seven ants, the honeybee, and various solitary insects to examine whether eusocial lineages share distinct features of genomic organization. Each ant lineage contains ∼4000 novel genes, but only 64 of these genes are conserved among all seven ants. Many gene families have been expanded in ants, notably those involved in chemical communication (e.g., desaturases and odorant receptors). Alignment of the ant genomes revealed reduced purifying selection compared with Drosophila without significantly reduced synteny. Correspondingly, ant genomes exhibit dramatic divergence of noncoding regulatory elements; however, extant conserved regions are enriched for novel noncoding RNAs and transcription factor–binding sites. Comparison of orthologous gene promoters between eusocial and solitary species revealed significant regulatory evolution in both cis (e.g., Creb) and trans (e.g., fork head) for nearly 2000 genes, many of which exhibit phenotypic plasticity. Our results emphasize that genomic changes can occur remarkably fast in ants, because two recently diverged leaf-cutter ant species exhibit faster accumulation of species-specific genes and greater divergence in regulatory elements compared with other ants or Drosophila. Thus, while the “socio-genomes” of ants and the honeybee are broadly characterized by a pervasive pattern of divergence in gene composition and regulation, they preserve lineage-specific regulatory features linked to eusociality. We propose that changes in gene regulation played a key role in the origins of insect eusociality, whereas changes in gene composition were more relevant for lineage-specific eusocial adaptations.
Genome Research (2013) gr.155408.113 -
The molecular clockwork of the fire ant Solenopsis invicta
[DOI]
[PDF]
K Ingram, C Moreau, A Kutowoi, Y Wurm, D Shoemaker, R Meier, G Bloch
The circadian clock is a core molecular mechanism that allows organisms to anticipate daily environmental changes and adapt the timing of behaviors to maximize efficiency. In social insects, the ability to maintain the appropriate temporal order is thought to improve colony efficiency and fitness. We used the newly sequenced fire ant (Solenopsis invicta) genome to characterize the first ant circadian clock. Our results reveal that the fire ant clock is similar to the clock of the honeybee, a social insect with an independent evolutionary origin of sociality. Gene trees for the eight core clock genes, period, cycle, clock, cryptochrome-m, timeout, vrille, par domain protein 1 & clockwork orange, show ant species grouping closely with honeybees and Nasonia wasps as an outgroup to the social Hymenoptera. Expression patterns for these genes suggest that the ant clock functions similar to the honeybee clock, with period and cry-m mRNA levels increasing during the night and cycle and clockwork orange mRNAs cycling approximately anti-phase to period. Gene models for five of these genes also parallel honeybee models. In particular, the single ant cryptochrome is an ortholog of the mammalian-type (cry-m), rather than Drosophila-like protein (cry-d). Additionally, we find a conserved VPIFAL C-tail region in clockwork orange shared by insects but absent in vertebrates. Overall, our characterization of the ant clock demonstrates that two social insect lineages, ants and bees, share a similar, mammalian-like circadian clock. This study represents the first characterization of clock genes in an ant and is a key step towards understanding socially-regulated plasticity in circadian rhythms by facilitating comparative studies on the organization of circadian clockwork.
PloS One (2012) 7:e45715 -
Epigenetics: the making of ant castes
[DOI]
[PDF]
A Chittka, Y Wurm, L Chittka°
Social insects represent a unique model for how the same genome can give rise to entirely different phenotypes — soldiers, common labourers, and queens. New research on ants and honeybees points to DNA methylation as a crucial factor in determining the caste of a developing individual.
Current Biology (2012) 22:R835-8 -
Visualization and quality assessment of de novo genome assemblies
[DOI]
[PDF]
O Riba-Grognuz°, L Keller, L Falquet, I Xenarios, Y Wurm°
Recent technological progress has greatly facilitated de novo genome sequencing. However, de novo assemblies consist in many pieces of contiguous sequence (contigs) arranged in thousands of scaffolds instead of small numbers of chromosomes. Confirming and improving the quality of such assemblies is critical for subsequent analysis. We present a method to evaluate genome scaffolding by aligning independently obtained transcriptome sequences to the genome and visually summarizing the alignments using the Cytoscape software. Applying this method to the genome of the red fire ant Solenopsis invicta allowed us to identify inconsistencies in 7%, confirm contig order in 20% and extend 16% of scaffolds.
Bioinformatics (2011) 27:3425-6 -
Relaxed selection is a precursor to the evolution of phenotypic plasticity
[DOI]
[PDF]
B Hunt, L Ometto, Y Wurm, D Shoemaker, S Yi, L Keller, M Goodisman°
Phenotypic plasticity allows organisms to produce alternative phenotypes under different conditions and represents one of the most important ways by which organisms adaptively respond to the environment. However, the relationship between phenotypic plasticity and molecular evolution remains poorly understood. We addressed this issue by investigating the evolution of genes associated with phenotypically plastic castes, sexes, and developmental stages of the fire ant Solenopsis invicta. We first determined if genes associated with phenotypic plasticity in S. invicta evolved at a rapid rate, as predicted under theoretical models. We found that genes differentially expressed between S. invicta castes, sexes, and developmental stages all exhibited elevated rates of evolution compared with ubiquitously expressed genes. We next investigated the evolutionary history of genes associated with the production of castes. Surprisingly, we found that orthologs of caste-biased genes in S. invicta and the social bee Apis mellifera evolved rapidly in lineages without castes. Thus, in contrast to some theoretical predictions, our results suggest that rapid rates of molecular evolution may not arise primarily as a consequence of phenotypic plasticity. Instead, genes evolving under relaxed purifying selection may more readily adopt new forms of biased expression during the evolution of alternate phenotypes. These results suggest that relaxed selective constraint on protein-coding genes is an important and underappreciated element in the evolutionary origin of phenotypic plasticity.
Proceedings of the National Academy of Sciences of the USA (2011) 108:15936-41 -
The genome of the leaf-cutting ant Acromyrmex echinatior suggests key adaptations to advanced social life and fungus farming
[DOI]
[PDF]
S Nygaard°, G Zhang, M Schiott, C Li, Y Wurm, H Hu, J Zhou, L Ji, F Qiu, M Rasmussen, H Pan, F Hauser, A Krogh, CJP Grimmelikhuijzen, J Wang, JJ Boomsma
We present a high-quality (>100× depth) Illumina genome sequence of the leaf-cutting ant Acromyrmex echinatior, a model species for symbiosis and reproductive conflict studies. We compare this genome with three previously sequenced genomes of ants from different subfamilies and focus our analyses on aspects of the genome likely to be associated with known evolutionary changes. The first is the specialized fungal diet of A. echinatior, where we find gene loss in the ant's arginine synthesis pathway, loss of detoxification genes, and expansion of a group of peptidase proteins. One of these is a unique ant-derived contribution to the fecal fluid, which otherwise consists of “garden manuring” fungal enzymes that are unaffected by ant digestion. The second is multiple mating of queens and ejaculate competition, which may be associated with a greatly expanded nardilysin-like peptidase gene family. The third is sex determination, where we could identify only a single homolog of the feminizer gene. As other ants and the honeybee have duplications of this gene, we hypothesize that this may partly explain the frequent production of diploid male larvae in A. echinatior. The fourth is the evolution of eusociality, where we find a highly conserved ant-specific profile of neuropeptide genes that may be related to caste determination. These first analyses of the A. echinatior genome indicate that considerable genetic changes are likely to have accompanied the transition from hunter-gathering to agricultural food production 50 million years ago, and the transition from single to multiple queen mating 10 million years ago.
Genome Research (2011) 21:1339-48 -
The genome of the fire ant Solenopsis invicta
[DOI]
[PDF]
Y Wurm°, J Wang, O Riba-Grognuz, M Corona, S Nygaard, B Hunt, K Ingram, L Falquet, M Nipitwattanaphon, D Gotzek, M Dijkstra, J Oettler, F Comtesse, C Shih, W Wu, C Yang, J Thomas, E Beaudoing, S Pradervand, V Flegel, E Cook, R Fabbretti, H Stockinger, L Long, W Farmerie, J Oakey, J Boomsma, P Pamilo, S Yi, J Heinze, M Goodisman, L Farinelli, K Harshman, N Hulo, L Cerutti, I Xenarios, D Shoemaker, L Keller
Ants have evolved very complex societies and are key ecosystem members. Some ants, such as the fire ant Solenopsis invicta, are also major pests. Here, we present a draft genome of S. invicta, assembled from Roche 454 and Illumina sequencing reads obtained from a focal haploid male and his brothers. We used comparative genomic methods to obtain insight into the unique features of the S. invicta genome. For example, we found that this genome harbors four adjacent copies of vitellogenin. A phylogenetic analysis revealed that an ancestral vitellogenin gene first underwent a duplication that was followed by possibly independent duplications of each of the daughter vitellogenins. The vitellogenin genes have undergone subfunctionalization with queen- and worker-specific expression, possibly reflecting differential selection acting on the queen and worker castes. Additionally, we identified more than 400 putative olfactory receptors of which at least 297 are intact. This represents the largest repertoire reported so far in insects. S. invicta also harbors an expansion of a specific family of lipid-processing genes, two putative orthologs to the transformer/feminizer sex differentiation gene, a functional DNA methylation system, and a single putative telomerase ortholog. EST data indicate that this S. invicta telomerase ortholog has at least four spliceforms that differ in their use of two sets of mutually exclusive exons. Some of these and other unique aspects of the fire ant genome are likely linked to the complex social behavior of this species.
Proceedings of the National Academy of Sciences of the USA (2011) 108:5679-84 -
Odorant binding proteins of the red imported fire ant, Solenopsis invicta: an example of the problems facing the analysis of widely divergent proteins
[DOI]
[PDF]
D Gotzek, C Moreau, H Robertson, Y Wurm, D Shoemaker
We describe the odorant binding proteins (OBPs) of the red imported fire ant, Solenopsis invicta, obtained from analyses of an EST library and separate 454 sequencing runs of two normalized cDNA libraries. We identified a total of 18 putative functional OBPs in this ant. A third of the fire ant OBPs are orthologs to honey bee OBPs. Another third of the OBPs belong to a lineage-specific expansion, which is a common feature of insect OBP evolution. Like other OBPs, the different fire ant OBPs share little sequence similarity (∼20%), rendering evolutionary analyses difficult. We discuss the resulting problems with sequence alignment, phylogenetic analysis, and tests of selection. As previously suggested, our results underscore the importance for careful exploration of the sensitivity to the effects of alignment methods for data comprising widely divergent sequences.
PloS One (2011) 6:e16289 -
 The genomic impact of 100 million years of social evolution in seven ant species
[DOI]
[PDF]
J Gadau°, M Helmkampf, S Nygaard, J Roux, D Simola, C Smith, G Suen, Y Wurm, C Smith
The genomic impact of 100 million years of social evolution in seven ant species
[DOI]
[PDF]
J Gadau°, M Helmkampf, S Nygaard, J Roux, D Simola, C Smith, G Suen, Y Wurm, C SmithAnts (Hymenoptera, Formicidae) represent one of the most successful eusocial taxa in terms of both their geographic distribution and species number. The publication of seven ant genomes within the past year was a quantum leap for socio- and ant genomics. The diversity of social organization in ants makes them excellent model organisms to study the evolution of social systems. Comparing the ant genomes with those of the honeybee, a lineage that evolved eusociality independently from ants, and solitary insects suggests that there are significant differences in key aspects of genome organization between social and solitary insects, as well as among ant species. Altogether, these seven ant genomes open exciting new research avenues and opportunities for understanding the genetic basis and regulation of social species, and adaptive complex systems in general.
Trends in Genetics (2011) 28:14-21 -
Behind the scenes of an ant genome project
[PDF]
Y Wurm
Formosan Entomologist (2011) 31:149-56 -
Parasitoid wasps: from natural history to genomic studies
[DOI]
[PDF]
Y Wurm, L Keller
The sequencing of three Nasonia genomes provides new insights on the molecular signature associated with parasitoid lifestyle, allows comparison with the social honey bee, and enables the identification of genes underlying between-species and sex-specific differences.
Current Biology (2010) 5:R242-4 -
Changes in reproductive roles are associated with changes in gene expression in fire ant queens
[DOI]
[PDF]
Y Wurm, J Wang, L Keller
In species with social hierarchies, the death of dominant individuals typically upheaves the social hierarchy and provides an opportunity for subordinate individuals to become reproductives. Such a phenomenon occurs in the monogyne form of the fire ant, Solenopsis invicta , where colonies typically contain a single wingless reproductive queen, thousands of workers and hundreds of winged nonreproductive virgin queens. Upon the death of the mother queen, many virgin queens shed their wings and initiate reproductive development instead of departing on a mating flight. Workers progressively execute almost all of them over the following weeks. To identify the molecular changes that occur in virgin queens as they perceive the loss of their mother queen and begin to compete for reproductive dominance, we collected virgin queens before the loss of their mother queen, 6 h after orphaning and 24 h after orphaning. Their RNA was extracted and hybridized against microarrays to examine the expression levels of approximately 10 000 genes. We identified 297 genes that were consistently differentially expressed after orphaning. These include genes that are putatively involved in the signalling and onset of reproductive development, as well as genes underlying major physiological changes in the young queens.
Molecular Ecology (2010) 19:1200-11 -
Fourmidable: a database for ant genomics
[DOI]
[PDF]
Y Wurm, P Uva, F Ricci, J Wang, S Jemielity, C Iseli, L Falquet, L Keller
The Fourmidable assembly pipeline groups nucleotide sequences into clusters before independently assembling each cluster. Subsequently, assembled sequences are annotated via Interproscan and BLAST against general and insect-specific databases. Gene-specific information can be retrieved using gene identifiers, searching for similar sequences or browsing through inferred Gene Ontology annotations. The database will readily scale as ultra-high throughput sequence data and sequences from additional species become available.
BMC Genomics (2009) 10:5 -
Sociogenetics of fire ants
[PDF]
Y Wurm
PhD Thesis; University of Lausanne, Switzerland (2009) -
Behavioral genomics: A, bee, C, G, T
[DOI]
[PDF]
Y Wurm, J Wang, L Keller
Honeybees, termites and ants occupy the ‘pinnacle of social evolution’ with societies of a complexity that rivals our own. The sequencing of the honeybee genome will provide a strong foundation for studying the genetic basis of complex social behavior.
Current Biology (2007) 17:R51-3 -
An annotated cDNA library and microarray for large-scale gene-expression studies in the ant Solenopsis invicta
[DOI]
[PDF]
J Wang, S Jemielity, P Uva, Y Wurm, J Graeff, L Keller
Ants display a range of fascinating behaviors, a remarkable level of intra-species phenotypic plasticity and many other interesting characteristics. Here we present a new tool to study the molecular mechanisms underlying these traits: a tentatively annotated expressed sequence tag (EST) resource for the fire ant Solenopsis invicta. From a normalized cDNA library we obtained 21,715 ESTs, which represent 11,864 putatively different transcripts with very diverse molecular functions. All ESTs were used to construct a cDNA microarray.
Genome Biology (2007) 8:R9 -
Cryostock a software for cell culture management
M Zerlin, L Greiffenberg, Y Wurm, I Winkler, D Kreusel, S Toemoe, L Ballabas, J Martin
During the last years the requirements on modern cell culture have rapidly increased, comprising quality control, global administration and global availability of cell lines. To meet these demands we developed CryoStock as a new software tool for cell culture management. This application combines a database for the storage of stocks and a LIMS for tracking all tasks during handling and transfer of cell lines. The data collected during the cell culture process can be used to follow parent/child relationships and to generate user friendly queries and reports. CryoStock will put another layer of quality on our cell culture handling to increase reproducibility and outcome of cell-based assays and to accelerate the knowledge transfer among Aventis' scientists.
Animal Cell Technology Meets Genomics (2005) 397-400