Part 3: Gene prediction
You need to have gone through Part 2: Genome assembly before starting this practical.
Many tools exist for gene prediction, some based on ab initio statistical models of what a protein-coding gene should look like, others that use similarity with protein-coding genes from other species, and others (such as Augustus and SNAP), that use both. There is no perfect tool or approach, thus we typically run many gene-finding tools and call a consensus between the different predicted gene models. MAKER and JAMg can do this for us. Let’s use MAKER on a sandbox example.
Running Maker
Create a new main directory for today’s practical (e.g., 2019-10-xx-gene_prediction) as well as the input, tmp, and results subdirectories and a WHATIDID.txt file to log your commands. Link the output (assembly) from yesterday’s practical into 2019-10-xx-gene_prediction/input:
cd ~/2019-10-xx-gene_prediction
ln -s ~/2019-09-xx-assembly/results/assembly.scafSeq input/
Pull out the longest few scaffolds from assembly.scafSeq into a new file:
seqtk seq -L 10000 input/assembly.scafSeq > tmp/min10000.fa
Next, cd to your tmp/ folder and run maker -OPTS. This will generate an empty maker_opts.ctl configuration file (ignore the warnings). Edit that file using a text editor such as nano or vim to specify:
- genome:
min10000.fa - augustus species: a known gene set from a related species, in this case we choose
honeybee1(yes that’s a 1) - deactivate RepeatMasker by replacing
model_org=alltomodel_org=(i.e., nothing) - further check that
repeat_proteinsetting is empty as well
For a real project, we would include RepeatMasker (perhaps after creating a new repeat library), we would provide as much relevant information as possible (e.g., RNAseq read mappings, transcriptome assembly – both improve gene prediction performance tremendously), and iteratively train gene prediction algorithms for our data including Augustus and SNAP.
Finally, run maker maker_opts.ctl. This may take a few minutes, depending on how much data you gave it.
Genome annotation software like MAKER usually provide information about the exon-intron structure of the genes (e.g., in GFF3 format), and sequence of corresponding messenger RNA and protein products (e.g., in FASTA format).
Once MAKER is done the results will be hidden in subdirectories of min10000.maker.output. MAKER provides a helper script to collect this hidden hidden output in one place (again please ignore the warnings for these steps):
# Pull out information about exon-intron structure of the predicted genes. This
# will be saved to the file min10000.all.gff.
gff3_merge -d min10000.maker.output/min10000_master_datastore_index.log
# Pull out predicted messenger RNA and protein sequences. These will be saved
# to the files: min10000.all.maker.augustus.transcripts.fasta, and
# min10000.all.maker.augustus.proteins.fasta
fasta_merge -d min10000.maker.output/min10000_master_datastore_index.log
Quality control of individual genes
So now we have some gene predictions… how can we know if they are any good? The easiest way to get a feel for this is to use the following example sequences: predicted protein sequences from rice and honeybee. We will compare them using BLAST to known sequences from other species in the uniref50 database.
Running BLAST
We will use SequenceServer to run BLAST. The command below will start a server. Once the server is ready, open http://localhost:4567 in your browser, paste the example rice and honeybee protein sequences in the textbox and click on the ‘BLAST’ button to run a BLAST search.
sequenceserver -d /import/teaching/bio/data/reference_databases/uniref50
Do any of the gene predictions have significant similarity to known sequences? For a given gene prediction, do you think it is complete, or can you infer from the BLAST alignments that something may be wrong? Start by comparing the length of your gene prediction to that of the BLAST hits. Is your gene prediction considerably longer or considerably shorter than BLAST hits? Why?
Now try a few of your gene predictions. BLAST only a maximum of 8 sequences at a time (instead of simply selecting the first 8 genes in your file, copy-paste sequences randomly from the file).
As you can see, gene prediction software is imperfect – this is even the case when using all available evidence. This is potentially costly for analyses that rely on gene predictions - i.e. many of the analyses we might want to do!
“Incorrect annotations [ie. gene identifications] poison every experiment that makes use of them. Worse still the poison spreads.” – Yandell & Ence (2012).
Using GeneValidator
The GeneValidator tool can help to evaluate the quality of a gene prediction by comparing features of a predicted gene to similar database sequences. This approach expects that similar sequences should for example be of similar length.
Although we won’t be running GeneValidator, please check the screenshots linked in the next sentence. Try to understand why some gene predictions have no reason for concern, while others do.
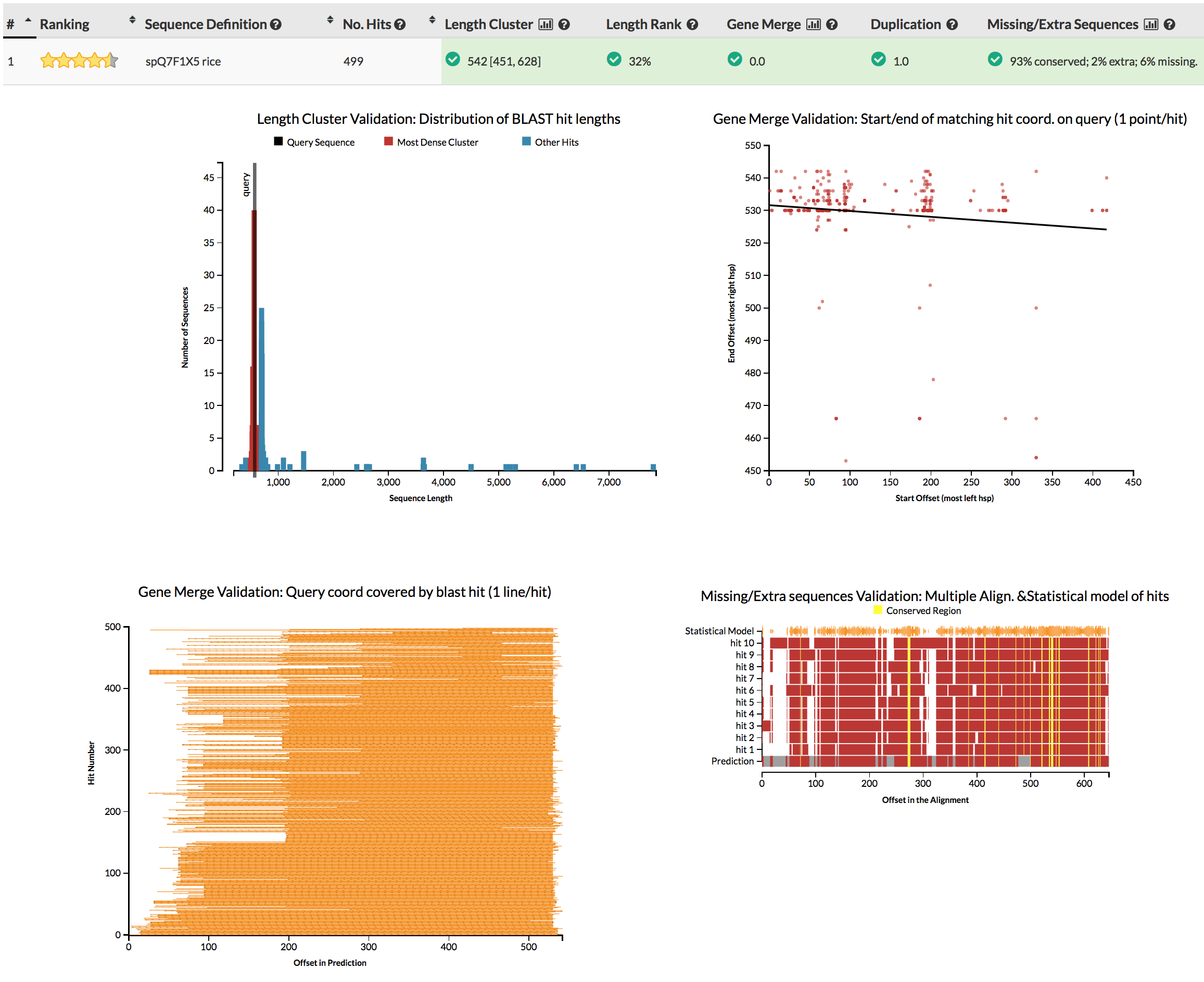{kind=link}
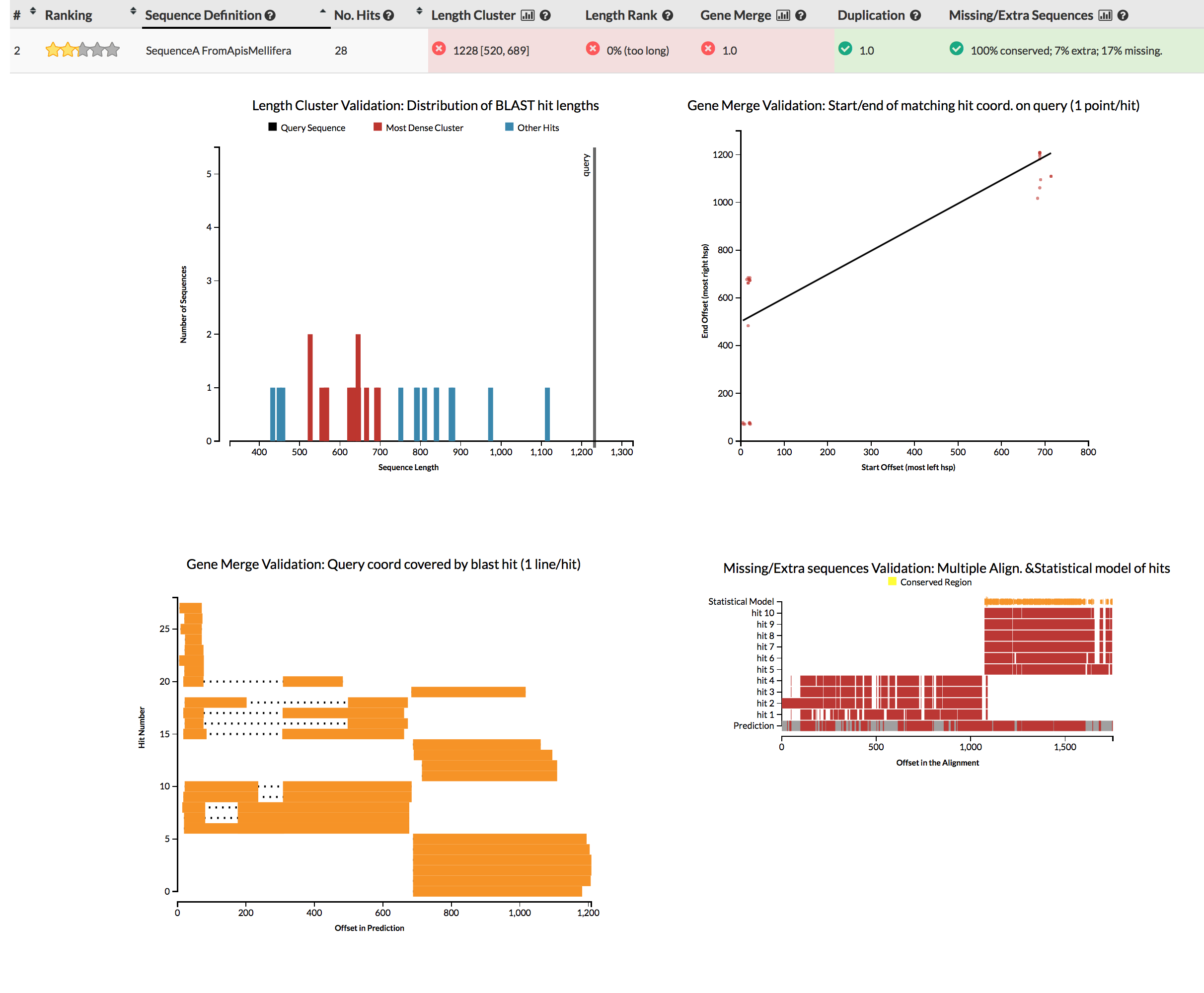{kind=link}
Comparing whole genesets & prioritizing genes for manual curation
Genevalidator’s visual output can be handy when looking at few genes. But the tool also provides tab-delimited output, useful when working in the command-line or running the software on whole proteomes. This can help analysis:
- In situations when you can choose between multiple gene sets.
- To identify which gene predictions are likely correct, and which predictions need might require further inspection and potentially be manually fixed.
Manual curation
Because automated gene predictions aren’t perfect, manual inspection and fixing is often required. The most commonly used software for this is Apollo/WebApollo.